Assistant professor Joseph Lyons coauthors milestone paper in Nature on lysosome function.
New article published in Nature entitled “ATP13A2 deficiency disrupts lysosomal polyamine export” sheds light on a defective lysosomal polyamine exporter (ATP13A2) that represents a lysosome-dependent cell death pathway that may be implicated in several neurodegenerative disorders including Kufor-Rakeb syndrome – a rare form of inherited juvenile-onset Parkinson’s disease.
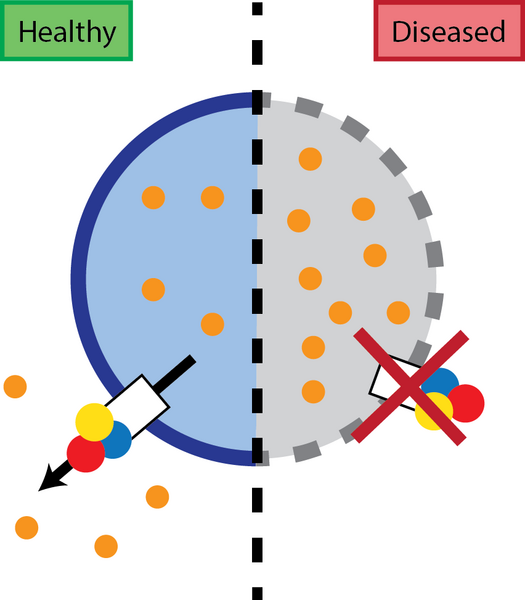
ATP13A2 belongs to a sub-family of P-type ATPases highly expressed in brain and neurons, whose function was previously unknown. Here, the researchers, led by Prof. Peter Vangheluwe (KU Leuven), found that ATP13A2 transports polyamines and is crucial for their uptake into the cell. Polyamines are physiologically important polycations that also play an important role in cellular homeostasis and are tightly regulated. They enter the cell via lysosomes where ATP13A2 exports these polyamines to the cell cytosol, a transport process essential for lysosomal function. Mutations in ATP13A2 affects the functionality of the transporter and cellular polyamine content, leading to polyamine accumulation in lysosomes, eventually leading to cell death.
In this paper researchers and co-author Joseph Lyons, from Prof. Poul Nissen’s laboratory at DANDRITE, demonstrate that impaired ATP13A2 function reduces late endo-lysosomal polyamine export, disrupting polyamine homeostasis and lysosomal function. Other recent genetic studies of Parkinson’s disease also suggest that lysosomal dysfunction contributes to the aggregation of alpha-synuclein and mitochondrial dysfunction leading to neuronal degeneration and thereby plays a major role in Parkinson’s disease.
Click here to read the publication.